Which of the following is the least correct statement regarding the DNA molecule?
The deoxyribose portion of the molecule encodes the genetic variation we see between different species.
How many possible reading frames do we need to evaluate when looking for genes in a DNA molecule?
6
Which of the following mutations is most likely to have the biggest phenotypic impact on an organism?
A in/del point mutation in a protein coding region.
Which of the following would be considered part of a gene?
- Introns
- Exons
- Start codon
What is the best estimate for the genetic distance between two sequences of DNA that have been allowed to mutate in an unrestrained manner while they have reached saturation?
0.75
Homologous sequences from two different species that fit the description in the question above would be a good source of data for phylogenetic analysis.
False
The simplest model of evolution we reviewed was the Jukes and Cantor (JC). Select the statement below that is false when considering this model of evolution.
This model should be used for all nuclear DNA data sets, while more complex models should be used for mitochondrial data sets.
What happens when a mutation results in a single amino acid substitution?
It can vary from a large impact (either good or bad) to no impact at all.
Protein domains ___.
often correspond to specific functional parts of the protein, like a transmembrane domain.
___ is the most common mechanism for the origin of new genes.
Gene duplication
Why was the original classification for Reptilia paraphyletic?
- Birds evolved a lot and now look very different from the ancestor of all reptiles (Sauria).
- Many of the lineages most closely related to the birds are now extinct.
Which of the following types of phylogenies shows both relationships among organisms and estimates rates of evolution along the branches?
Phylogram
Neighbor-joining is a method of phylogenetic inference that uses ___ data and ___.
Distance, clustering algorithm
Xenology is most common among ___.
Prokaryotes
Using the genetic matrix below and the principle of parsimony, which of the trees is the best hypothesis for relationships among the species?
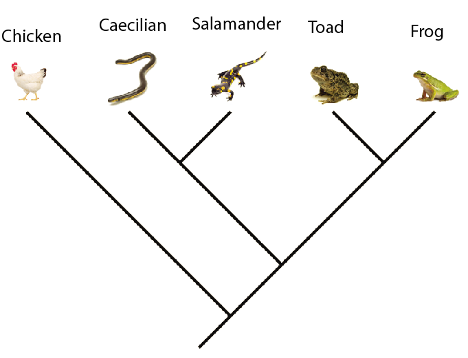
On the best phylogeny above, what is the closest relative of the Caecilian?
Salamander
Which of the characters above provides no evidence for relationships among the various species?
Character 8
Classifying the caecilian, toad, frog, and salamander together would be a valid group based on the best phylogeny above.
True
Are the lysozymes that aid in cellulose digestion in the cow and the langur monkey homologous?
Yes, as are the corresponding lysozymes in humans, however we know based on relationships of these organisms that the ability to digest cellulose was gained independently in these organisms.
What information about protein coding genes might help when we try to align them?
Due to the degeneracy of the genetic code they are more conserved at the amino acid level.
Parsimony
This method treats all mutations equally and uses an optimality criterion of the phylogeny that minimizes the number of overall mutations.
Maximum likelihood
A method that used discrete data, an optimality criterion, and models of evoluation.
Minimum evolution
A distance method that has an optimization criterion but is not frequently used anymore.
Which of the following is an advantage of maximum likelihood over parsimony?
It allows you to perform statistical test to see if one phylogeny is significantly better than another.
Optimality methods of phylogenetic inference are NP-Complete, this means ___.
there is no algorithm to find the optimal solution for relatively small data sets (50 species) and we can't evaluate every possible solution.
What is a gene?
A section of DNA that is transcribed into RNA and has a phenotypic impact.
What is the difference between a purine and a pyrimidine?
Purines (adenine and guanine) have two carbon rings, pyrimidines (cytosine and thymine) have one.
What is the disadvantage of choosing more complex model of evolution?
More complexity means a loss of precision due to the estimation of many different parameters.
Why does denaturation have such a profound effect on protein function?
A denatured protein has lost its 3D shape, since shape is critical for function this means denatured proteins cannot perform their function.
The diagram below is a data set similar to the one you worked on for project 1. What does a row and a column in this data set represent?
- Row: DNA sequence from a representative species
- Column: homology statement (aligned so each position in all genes match up)
What are the two things that can be represented by a polytomy?
1. Uncertainty about relationships.
2. Simultaneous divergence of three or more descendant lineages.
What is the best way to determine of two similar characteristics in different species are homologous?
Map the origin of that trait onto a phylogeny.
What are two sources of homoplasy in a phylogenetic analysis?
1. Convergence
2. Symplesiomorphy
List the main advantage and one disadvantage of neighbor joining.
- They are very efficient (fast, little computational time).
- Disadvantage: distance data instead of discrete, no optimization criterion, often not as accurate as more inefficient methods.
What does it mean when we say Maximum Likelihood analyses are circular?
They require a model of evolution but to pick the model and to estimate the parameters for this model we need a phylogeny.
Define paralogy and describe a specific example.
Paralogy is when two homologous genes can trace their ancestral gene to a gene duplication event. Any two members of a gene family could serve as a specific example, like the hoatzin and mammal lysozymes we talked about in class, alpha and beta hemoglobin proteins, and many others.
We can never be 100% certain that we've found the true phylogeny for a group of organisms, but what are two approaches that might give us confidence that our methods finding the best phylogenies are actually valid?
1. Testing our method on known phylogenies or simulated data sets for which we know the relationships.
2. Congruence of methods: if more than one methodology yields identical (or very similar) phylogenies we have more confidence that our phylogenies represent the actual species history.
I complete a phylogeny of reptiles and want to be 100% sure it's completely accurate. Which of the following statements is most correct about my process of ensuring 100% accuracy?
Although congruence among methods might increase by confidence that mu results are accurate, because I am trying to reconstruct historical relationships, I can never know with 100% certainty that my results are accurate.
If I accurately reconstruct the history of genes, I will always also reconstruct the history of the species to which those genes belong.
False
What do the numbers above nodes represent in Bayesian analysis?
These are posterior probability scores estimated via a majority rule consensus tree of all phylogenies generated after the "burn-in" period.
The Markov Chain Monte Carlo (MCMC) method allows for a rapid search of nearby phylogenetic tree space during a Bayesian analysis allowing for eventual optimization of a model of evolution and its parameters.
True
What is meany by the term "congruence" when referring to phylogenetic trees?
That the patterns of relationships in the final phylogenies are completely or largely the same.
Bootstrap support
Requires the creation of new data sets by sampling the original data matrix with character replacement.
Jackknife support
Helps to determine how sensitive a data set is to taxon sampling.
Posterior probability
Is created by making a consensus tree from all the phylogenies stored during the post "burn-in" phase of a Bayesian analysis.
Although bootstrap support values are widely used there is good evidence that they tend to overestimate levels of confidence in relationships.
True
Which of the following is not a valid criticism of supertree methods?
Models of evolution cannot be incorporated in any of the components of a supertree analysis.
Which of the following best describes why researchers first began to use a supertree approach?
They were originally created as a way to combine results from separate analyses where the underlying data was not congruent enough to assemble into a single data set.
Which of the following supertree methods is the most efficient?
Informal
Both symplesiomorphy and convergence represent a type of homoplasy and therefore represent a less parsimonious pattern of evolution than synapomorphies.
True
This supertree method uses an initial phylogeny (ex: a phylogeny generated via neighbor-joining) and then defines several parts of the overall tree with shared taxa between parts. Data sets for each of these parts are then generated and individual analyses of the parts are combined using an optimization method.
Disk covering method
Select all of the answers below that accurately describes a pattern of characters mapped onto the phylogeny below.
- Character Z is a symplesiomorphy for the group consisting of species D, E, F, and G
- Character Y provides no evidence for relationships between these species
- Character X is best categorized as convergence
When two distantly related lineages gain a similar feature it is called ___.
convergence
Fitch optimization is a method that allows for a most parsimonious mapping of complex character patterns onto any given phylogeny.
True
Which of the following is not a method that would be useful in testing alternative phylogenetic hypotheses?
Posterior branch support
Which of the following is the most accurate statement regarding the mapping of characters onto a phylogeny?
It allows for the estimation of ancient ancestral character states, even if we have no fossil evidence for that ancestor.
Which of the following best describes the mapping of character states on to ancestral nodes in a maximum likelihood analysis?
Character states are represented by probabilities at each node, these probabilities are calculated using the model of evolution and the relative branch lengths.
The likelihood ratio test (LRT) is a very flexible statistical test and can be used to determine if there is a significant difference between many different types of molecular analyses. Which of the following is not an application of the LRT?
Test the difference between different competing equally parsimonious fitch optimization character mappings.
Anagenesis corresponds with which of the following?
Length of internal branches
Cladogenesis corresponds with which of the following?
Speciation events
If I was doing a phylogeny of species using genes that were all part of a large gene family which of the following would give me the best chance of accurately reconstructing the species history?
Identify orthologous gene copies and represent each orthologous set of genes as a separate component of the overall matrix.
Which of the following is the most widely used species concept?
Morphological
What does it mean when we say a phylogenetic hypothesis is precise?
How many other answers are just as good, or similar enough so that they are not statistically different than the best answer.
How is a Bremer support value calculated?
The difference between the score of most parsimonious tree and the score of the best tree that doesn’t contain a particular node.
What is the effect of poor taxon sampling on a phylogeny?
This creates large gaps (longer branch lengths) and can compromise the accuracy of results.
What is an informal supertree?
A tree made from the result of other analyses using a “copy and paste” method with no formal analysis.
What is the biological process that creates xenology?
Horizontal gene transfer
What is Dollo parsimony and what type of characters is it best applied to?
A form of weighted parsimony, a trait may be lost multiple times, but never re-evolves. This applies best to complex morphological characters.
How does one select the best model of evolution to use for a Maximum Likelihood analysis?
A likelihood score can be generated for each model and then the best fit model can be determined using the Likelihood Ratio Test.
What is a majority rule consensus tree?
A tree that summarizes relationships of two or more phylogenies, numbers above nodes represent the frequency of those nodes in the constituent trees.
What is different about reconstructing the phylogenetic history of bacteria compared to eukaryotic species?
Because of horizontal gene transfer relationships among bacteria are more like a network than a tree.
Why is there no single definition for a species?
Because speciation is a process and different factors can influence both how and how fast a species breaks into descendant lineages.
What was the main impetus for the development of Bayesian approaches to phylogenetics and how is it different than Maximum Likelihood approaches?
Researchers wanted to be able to use models of evolution and statistical approaches to estimate phylogenetic relations, but Maximum Likelihood methods were very slow. Bayesian estimations calculate the likelihood score differently and generate support values (posterior probabilities) as a part of the initial analysis.
Define lineage sorting and list two evolutionary factors that make it more likely.
When allelic diversity is maintained for long periods of time and subsequent fixation of alleles in descendant lineages creates a gene history that doesn’t match the species history. Rapid speciation events and long coalescent times increase its likelihood.
Which of the following would be the best approach to estimate the Heterozygosity of a population?
Genotype a randomly selected sample of the population for a number of different loci.
Why does a small population size create a population that is not at HW equilibrium?
- It causes the effects of genetic drift to be greater
You survey a population of wild cheetahs and find the following distribution of genotypes: AA: 360 Aa: 480 aa: 160 What are the allele frequencies for this population and are there evolutionary forces acting on this gene?
p = 0.6 q = 0.4, no sign of evolutionary forces
You sample a gene in a chimpanzee population with the following results: AA: 300 Aa: 600 aa: 100 Which of the following statements about this gene is the most accurate?
There is an evolutionary force acting on this gene, if it is natural selection it is most likely overdominance
Ka/Ks > 1
A venom gene that has evolved to specifically target different prey items of the different sampled species
Ka/Ks < 1
NADH2 a gene critical for metabolism with identical function in all sampled species
Ka/Ks = 1
A pigmentation gene that is expressed in the dermis, but has no impact at all on the fitness of any of the sampled species
Much of the diversity present within a population arose due to mutations that change only one base pair. What is this type of diversity called?
Single Nucleotide Polymorphism (SNP)
One of the fastest evolving types of genetic makers are made up of tandem repeat of DNA that mutate to have different numbers of repeats. What is this type of diversity called?
Microsatellite
Most genes in wild populations
are polymorphic
Which of the following genes in humans is least likely to be subject to recombination?
NADH2, found on the mitochondrion
What two values would I need to estimate theta (Q)?
- Population size
- Mutation rate
The vast majority of genetic diversity in the human population shows no correlation with geography.
True
Fst is a measure of genetic structure within a population. Which of the following describes how this value is determined?
It is estimated by looking for differences in heterozygosity in subpopulations when compared to the total population.
When formulating his theory of natural selection Darwin was evaluating ________, however the majority of genetic diversity _________.
diversity that changed phenotype, is neutral
If natural selection works to remove deleterious mutations, why do we still see them in populations?
A number of factors including genetics, environmental instability and varying strengths of selection means that the removal of some deleterious mutations is very slow.
What accounts for the majority of the C-value paradox?
The differing amounts of non-coding DNA in eukaryotic genomes
Which of the following is the best way to calibrate the molecular clock?
Use fossil data (when available) to get an independent estimate of coalescent times.
What is the primary reason to perform a relative rate test?
To determine whether a molecular clock can be assumed for the data set being analyzed.
These proteins can all be classified as part of a single protein family.
Opsins
Co-option of a suitable subset of genes that perform a useful physiological function is the major force explaining their diversity.
Venom
These proteins are found primarily on the mitochondrial genome.
None of the above
Which of the following is the most accurate statement regarding genomes and transcriptomes.
Mapping a complete transcriptome onto a genome is the best way of determining the loci of all genes in the genome.
Which of the main types of RNA are we targeting when doing transcriptome sequencing?
mRNA
“Microevolution” and “macroevolution” are caused by different evolutionary forces.
False
What is effective population size (Ne)? How is this different than census population size (N)?
Census population is an estimate of all individuals in a population, however effective population size is an estimate of the number of individuals that will contribute to the next generation.
List two things that distinguish the mitochondrial genome from the nuclear genome.
- Circular
- Less coding DNA
- Ancestrally was gained via endosymbiosis of bacteria
- Only inherited from the mother
- Different number of copies than the nuclear genome
What is the relative rate test?
An estimate of the genetic distance between two separate pairs of species that all share the same common ancestor. This can help us to know if the data can be used to estimate divergence time for these species.
What effect does natural selection have on neutral mutations?
By definition, natural selection has no effect on neutral mutations (only beneficial and detrimental mutations).
What happens to brand new mutations that are beneficial? List two factors that might influence this.
These mutations will spread through the population generation after generation. The rate of this spread can be influenced by genetics, strength of the benefit of the new mutation, stability of the environment that determines its advantage and the life stage at which the mutation provides a benefit.
Why are dN/dS (Ka/Ks) values often elevated after a recent zoonotic transmission event?
This reflects the selective pressure on populations due to the changed environment of the new host/parasite interaction.
What are GC content bias patterns like in prokaryotes?
They can vary widely between species ( ~25% - ~85%) but tend to be much across the genome of a single species.
What three metazoan groups have evolved complex eyes and vision?
1. Arthropods
2. Vertebrates
3. Mollusks
What is pleiotropy and how does this relate to proteins that act as venoms?
Pleiotropy is when one gene has more than one phenotypic impact. After recruitment many proteins used as venoms maintain their original function, this means there’s a higher amount of evolutionary constraint due to the two separate jobs for this protein.
What is the G-value paradox and what are the three explanations for this paradox?
- Changes to non-coding regions can create complex gene regulation
- Complex network interactions
- Some genes have multiple protein products (differential splicing)
List four reasons why most mutations can be considered as neutral.
- large amounts of eukaryotic genomes are non-protein coding
- degeneracy of DNA code
- some mutations change the phenotype, but have insignificant impact on fitness
- some mutations change the amino acid, but the protein still functions in the same way
Outline how a Quantitative Trait Loci (QTL analysis) is performed.
An analysis is performed to look for linkage disequilibrium for any genetic condition and one or more of the tens of thousands of genetic markers in the human population. High levels of linkage disequilbrium in different parts of the genome don’t indicate a specific cause for the condition, but indicate that there is something in that area of the genome that contributes to the condition.