Instructions for Side by Side Printing
- Print the notecards
- Fold each page in half along the solid vertical line
- Cut out the notecards by cutting along each horizontal dotted line
- Optional: Glue, tape or staple the ends of each notecard together
Unit 11 Heme Derivatives
front 1 What is heme comprised of ? | back 1 Protoporphyrin Ring + Fe |
front 2 What is Hemoglobin comprised of ? | back 2 Heme + 4 globin chains there are many possible chains, but the common ones are alpha, beta, delta, and gamma] |
front 3 What is the problem with unbound iron? | back 3 it is toxic to human tissues |
front 4 What is the primary purpose of Ferritin? | back 4 it is the storage form of iron and made up of the Ferric (3+) form, which is stored in the liver |
front 5 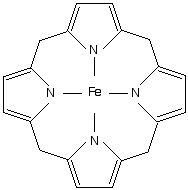 What is the definition of Porphyrins? | back 5 any of a class of pigments (including heme and chlorophyl) whose molecules contain a flat ring of four linked heterocyclic groups, sometimes with a central metal atom. |
front 6 What is the definition of Porphyrias? | back 6 It is named from the ancient Greek word porphura, meaning purple. These diseases are caused by enzyme deficiencies in the heme production pathway. People generally look purple when they have this disease. |
front 7  What is the definition of Hemin? | back 7 - a black inorganic compound which forms from heme when RBCs are lysed and exposed to air, Can be used as treatment for some porphyrias |
front 8 What is the definition of Porphyria Cutanea Tarda ? | back 8 Deficiency of Uroporphyrinogen Decarboxylase defined by blistering (most common) |
front 9 What test is used for Porphyria Cutanea Tarda? | back 9 serum porphyrin is the best test to order |
front 10 What is the definition of Acute Intermittent Porphyria? | back 10 Deficiency of PBG Deaminase, defined by blistering and nerve impairments (2nd most common) |
front 11 What test is used for Acute Intermittent Porphyria? | back 11 urine porphobilinogen is the best test to order |
front 12 How do you diagnosis Erythropoietic Porphyria? | back 12 it must show an increase in total erythrocyte protoporphyrin |
front 13 What is the definition of Erythropoietic Porphyria? | back 13 - Deficiency of ferrochelatase defined by blistering |
front 14 What test is used for ALA-Dehydratase Deficiency? | back 14 the Delta-aminolevulinic acid assay |
front 15 What is important to know about ALA-Dehydratase Deficiency? | back 15 it is important because it’s the rate-limiting step in porphyrin synthesis |
front 16 What is the 2nd most common type of porphyria? | back 16 Acute Intermittent Porphyria (AIP) is the second most common porphyria. Patients suffer acute neurological disease and urine turns brown with exposure to light. |
front 17 What does the following common secondary porphyrias do: Lead poisoning? | back 17 inhibition of most of the enzymes in the synthetic pathway |
front 18 What does the following common secondary porphyrias do: Hereditary tyrosinemia ? | back 18 elevated levels of succinylacetone which inhibits Aminolevulinic Acid Dehydratase (ALAD) resulting in elevated levels of d-Aminolevulinic Acid |
front 19 What does the following common secondary porphyrias do:Liver disease - | back 19 leads to a problem synthesizing heme |
front 20 What does the following common secondary porphyrias do: Iron deficiency anemia - | back 20 leads to an increase in heme production, therefore more precursor substances are needed, potentially leading to a porphyria. Erythrocyte protoporphyrin is increased in these patients. |
front 21 What are the expected lab values for Iron Deficiency Anemia? | back 21 Iron: Low Transferrin: High Transferrin Saturation: Low Total Iron Binding Capacity: High Ferritin: Low |
front 22 What is the Porphobilinogen Methods of Analysis? | back 22 Watson-Schwartz - 4-dimethylaminobenzaldehyde (Ehrlich’s reagent) produces a red color in the presence of porphobilinogen which is detected at 555 nm spectrophotometrically. |
front 23 Porphyrins are evaluated using primarily? | back 23 HPLC with fluorescent detection note: We typically assess plasma, urine, and stool for porphyrins, using chromatographic analysis (with HPLC) All of the samples MUST be protected from light |
front 24 What is Porphobilinogen (PBG) Testing used for? | back 24 to confirm Acute Intermittent Porphyria (AIP) or to evaluate disease risk in patients with direct family members with AIP |
front 25 What does Spectrofluorometric assay use? | back 25 porphobilinogen deaminase MEMORY TRICK: Think “AIPBG”, the disease together with the diagnostic test |
front 26 what is Ferrochelatase Testing used for? | back 26 screening test for Lead poisoning |
front 27 How long does it take for Senescent” erythrocytes to die ? | back 27 around 120 days note: Heme is either reused/recycled OR metabolized into bilirubin and excreted |
front 28 What does Unconjugated Bilirubin = | back 28 Indirect Bilirubin |
front 29 what does Conjugated bilirubin = | back 29 Direct bilirubin (hydrophilic) |
front 30 What is Crigler-Najjar or Gilbert syndromes? | back 30 Increases in unconjugated bilirubin occur with hemolysis or inherited liver disease |
front 31 What is Dubin-Johnson or Rotor syndromes? | back 31 Increases in conjugated bilirubin occur with biliary obstruction (tumor) or inherited liver disease |
front 32 What does Delta Bilirubin = | back 32 Conjugated bilirubin covalently bound to albumin |
front 33 What does Total Bilirubin = | back 33 Conjugated + Unconjugated + Delta Bilirubin |
front 34 What is the common name for unconjugated bilirubin? | back 34 Indirect bilirubin |
front 35 What are the serious autosomal dominant disorders? | back 35 1.Dubin-Johnson Syndrome 2.Crigler-Najjar Syndrome |
front 36 What are the Weak sauce autosomal recessive disorders? | back 36 1.Rotor Syndrome 2.Gilbert Syndrome |
front 37 What are the Conjugated hyperbilirubinemia disorders? | back 37 Dubin-Johnson Syndrome Rotor Syndrome |
front 38 What are the Unconjugated hyperbilirubinemia? | back 38 Crigler-Najjar Syndrome Gilbert Syndrome |
front 39 What would you expect the bilirubin levels to look like in a patient with acute cholecocholithiasis? | back 39 Indirect bilirubin high Direct bilirubin high |
front 40 What is the absorption of the Evelyn-Malloy Method? | back 40 Creates a red-purple chromogen with maximal absorption at 560 nm |
front 41 What is the absorption of the Jendrassik-Grof Method? | back 41 Creates a blue chromogen with maximal absorption at 600 nm |
front 42 What are the Preanalytical Concerns for Bilirubin Analysis? | back 42 A fasting sample is preferred to reduce alcohol interference Lipemia will falsely increase the measured bilirubin Hemolyzed samples will decrease the reaction with the diazo reagent Bilirubin is very light sensitive and may be 50% destroyed in one hour of exposure |
front 43 What are the optimal storage conditions for Bilirubin Analysis? | back 43 when the serum or plasma is separated from the cells, stored in the dark and for up to 2 days at RT, 1 week at 4˚C, and indefinitely at -20˚C. |
front 44 What are the steps for Hemoglobin Electrophoresis ? | back 44 First performed on cellulose acetate using an alkaline buffer (pH 8) Secondarily performed on an acid buffer (pH 6) to distinguish hemoglobins note: The order of hemoglobins from slowest to fastest is C, S, F, and A |
front 45 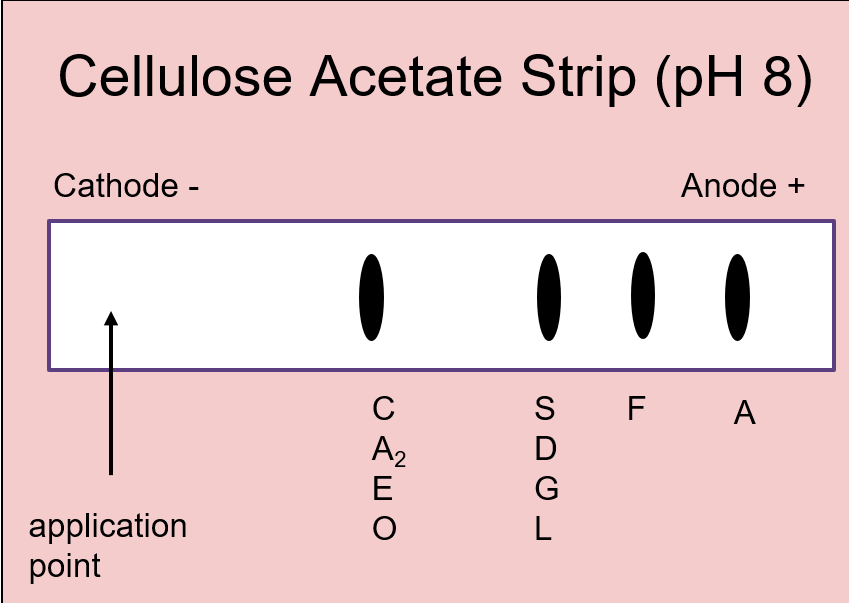 What is the purpose of Alkaline strips in hemoglobin? | back 45 to detect abnormal hemoglobin's |
front 46 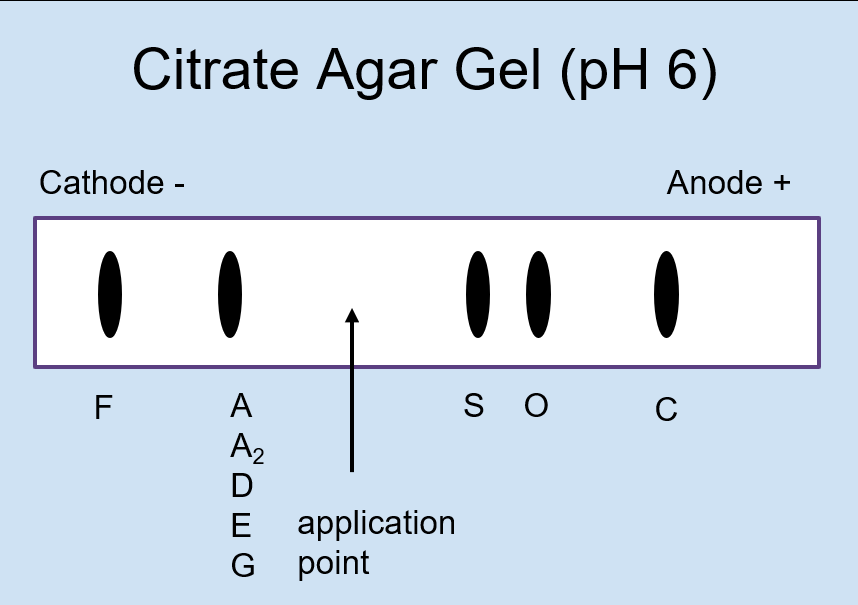 What is the purpose of Acid in hemoglobin Electrophoresis? | back 46 to differentiate abnormal hemoglobins that migrate together on alkaline hemoglobin electrophoresis |
front 47 Why do hemoglobins S, D, and G migrate in the same position on an alkaline hemoglobin electrohporesis? | back 47 The side chains of the abnormally substituted amino acids in the hemoglobin react similarly at a pH of 8.4. The same goes for the group of hemoglobins C, A2, E, and O. |
front 48 What is the difference between Hemoglobin E vs A2? | back 48 Hemoglobin A2 is normal but Hemoglobin E has a mutated beta-chain. To distinguish, perform capillary electrophoresis |
front 49 What is the Abnormal Acquired Hemoglobin: HbA 1c? | back 49 glycated hemoglobin, used for monitoring glucose control in diabetic patients |
front 50 What is the Abnormal Acquired Hemoglobin:Carboxyhemoglobin ? | back 50 - carbon monoxide binds to hemoglobin and displaces oxygen and binds with greater affinity, seen in house fires, enclosed areas near an exhaust source, etc. |
front 51 What is the Abnormal Acquired Hemoglobin: Methemoglobin? | back 51 Alkaline media or oxidizing agents transform normal Fe2+ into Fe3+, conferring a inability to reversibly bind oxygen |
front 52 What is the Abnormal Acquired Hemoglobin: Sulfhemoglobin ? | back 52 - Sulfation of hemoglobin causes an inability to bind oxygen |
front 53 What is the Iron Spectrophotometry Method? | back 53 1.Release iron from transferrin with addition of acid 2.Reduction of Fe3+ to Fe2+ with ascorbic acid 3.React Fe2+ with a chromogen (e.g. ferrozine) |
front 54 What percent of iron binds to carrier proteins? | back 54 ⅔, or 66% |
front 55 Iron Deficiency - Lots of transferrin, very little iron = | back 55 ↑TIBC ↓Fe |
front 56 Anemia of Chronic Disease - Plenty of iron, out of circulation = | back 56 ↓TIBC ↓Fe |
front 57 Hemochromatosis - Defective transporter cannot remove iron = | back 57 ↓ TIBC ↑Fe |
front 58 What is Hemoglobin H Disease? | back 58 Inadequate alpha globulin production Produces Beta Hemoglobin tetramers |
front 59 What 4 globin chains make up hemoglobin A1? | back 59 2 alpha and 2 beta |
front 60 what the difference between a heme ring and a protoporphyrin IX ring is? | back 60 Think of Protoporphyrin IX as being Tony |
front 61 What compounds serve as the negative feedback mechanism for ALA synthase? | back 61 Glucose and Heme |
front 62 What is the 2nd most common form of porphyria? | back 62 Acute Intermittent Porphyria (AIP) is the 2nd most common porphyria. Patients suffer acute neurological disease and urine turns brown with exposure to light. |
front 63 what if I had a patient that was producing normal hemoglobins,
but | back 63 We call these thalassemias |
front 64 If you have the following alleles what is the disease state: β/ β+ or β/ β0 | back 64 β thalassemia minor (clinically silent) |
front 65 If you have the following alleles what is the disease state: β+/β0 or β+/β+ | back 65 β thalassemia intermedia (moderate) |
front 66 If you have the following alleles what is the disease state: β0/β0 | back 66 β thalassemia major (severe) |
front 67 βThalassemia is most often caused by what type of mutation? | back 67 Point mutation in the β globin gene |
front 68 A potential mother comes into the genetic counselor’s office asking regarding her future child’s risk for | back 68 Her child would have a 1 in 4 chance of having |
front 69 A 46-year-old female presents to her physician with possible sickle
cell trait, as both of her parents were carriers. Her physician orders
a hemoglobin electrophoresis. What phenotypic results would show that
she is a carrier for sickle cell trait? | back 69 The correct answer is A, HbA2/S. This is correct because he would
have received one normal gene producing HbA2 and one abnormal gene
producing HbS. This genotype is considered a “carrier” of the
mutation, which may or may not significantly affect their lives. B.
HbC/S disease can be quite detrimental to the carriers of both HbC and
HbS genes. The red cells in these patients still undergo sickling
under hypoxic stress just like in sickle cell disease. HbF/S would be
a normal finding in sickle cell disease because the production of HbF
is increased in order for the body to be able to carry some
more |
front 70 A 2-day-old male infant is taken to his pediatrician by his mother. During a thorough history and physical, it was revealed that Hemoglobin H disease runs in the family. His current signs include a Hb of 9 g/dL and MCV of 70 fl. What hemoglobin chains constitute Hemoglobin H? A. 2 α 2δ, B. 2 α , 2β C. 2 α , 2 γ D. 4 β | back 70 The correct answer is D) 4β chains. Very few α chains are produced in α -thalassemia with 3 out of 4 α -globin chains defective (HbH disease) and no α chains are produced when all 4 chains are defective, and is incompatible with life (hydrops fetalis). Because there are not enough α chains to bind, the β chains just go ahead and form tetramers which are called HbH. A) is the normal variant HbA2 , B) is the normal HbA, and C) is HbF. |
front 71 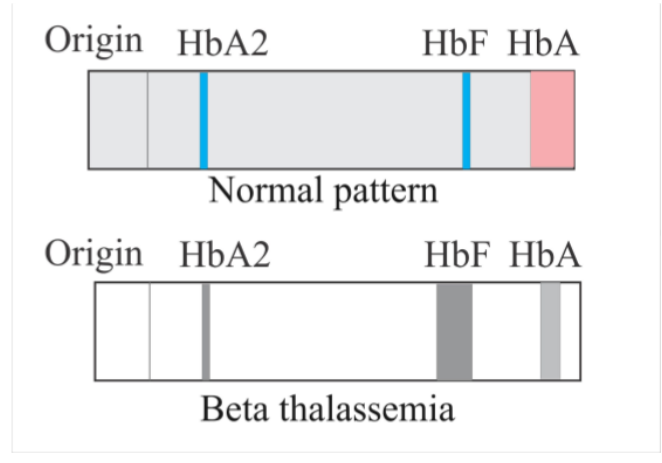 A 43-year-old female with β-thalassemia intermedia presents to her primary care physician for management of her disease. A hemoglobin electrophoresis is ordered. How would β-thalassemia affect the HbF and HbA bands on the electrophoresis? | back 71 The correct answer is B) Decreased HbA and Increased HbF. Let’s dive
into why. Normally, we have a lot of HbA, a little HbA2 and adults
have very little HbF. These hemoglobins are made up of different
hemoglobin chains. HbA is made up of 2α chains and 2β chains and HbF
is made up of 2α chains and 2γ chains. In β-thalassemia, the
β |
front 72 What should you associate “coproporphyrinogen III” with? | back 72 heme synthesis |
front 73 What hemoglobins should you know? | back 73 HbA, HbA2 ,HbS, and HbF. Remember that HbA2 crawls with HbC. |
front 74 What is the Enzyme Deficiency for:PBG Synthgase (AKA ALA Dehydratase) | back 74 ALA Dehydratase |
front 75 What is the Enzyme Deficiency for: Uroporphobiliogen III Cosynthase | back 75 Congenital Erythropoietic Porphyria |
front 76 What is the Enzyme Deficiency for: Uroporphobilinogen Decarboxylase | back 76 Porphyria Cutanea Tarda |
front 77 What is the Enzyme Deficiency for: Ferrochelatase | back 77 Erythropoietic porphyria |